
A team led by scientists Prof. Thomas D. Kühne and Prof. Christian Plessl at Paderborn University has broken the exascale barrier for mixed-precision computation for a computational science application and has achieved 1.1 EFLOP/s application-level performance in the quantum chemistry application CP2K on the NERSC Perlmutter system. The results are reported in a pre-print that has just been published:
Robert Schade, Tobias Kenter, Hossam Elgabarty, Michael Lass, Thomas D. Kühne, Christian Plessl
Breaking the Exascale Barrier for the Electronic Structure Problem in Ab-Initio Molecular Dynamics
Preprint: https://doi.org/10.48550/arXiv.2205.12182
Currently, we are on the cusp of the exascale era and it is widely expected that the first supercomputer to break the exascale threshold for double precision floating-point computations will be publicly announced at the ISC High-Performance Conference (ISC) in Hamburg in late May. This milestone will mark the end of a race to exascale that has considerably heated up with globally increased competition for scientific leadership and can been rightly referred to as the "Space Race of the 21st Century."
The merits and deficits of the HPL benchmark to rank the most powerful supercomputers has been widely discussed. Any HPC practitioner knows that extracting anything close to HPL performance and efficiency is very ambitious and that many scientific codes exploit only a small fraction of the theoretical performance due to limited parallelism, insufficient vectorization opportunities, communication overheads, load imbalance, etc. Thus, practically exploiting the capabilities of an exascale computer for computational science will require adaptations to algorithms, numerical libraries and application codes that have been initiated a few years ago.
New method for massively parallel quantum chemistry simulation
The team in Paderborn has taken up the challenge with regard to exascale computing for the field of quantum chemistry and has developed a novel method of which the initial variant has been presented at the SC’20 conference (Lass et. al: Submatrix Method for the Approximate Computation of Matrix Functions, https://doi.org/10.5555/3433701.3433807). The method has been advanced by the authors in 2021 to a largely improved highly scalable and efficient method with an efficient GPU acceleration (Schade et. al: Towards electronic structure-based ab-initio molecular dynamics simulations with hundreds of millions of atoms, https://doi.org/10.1016/j.parco.2022.102920).
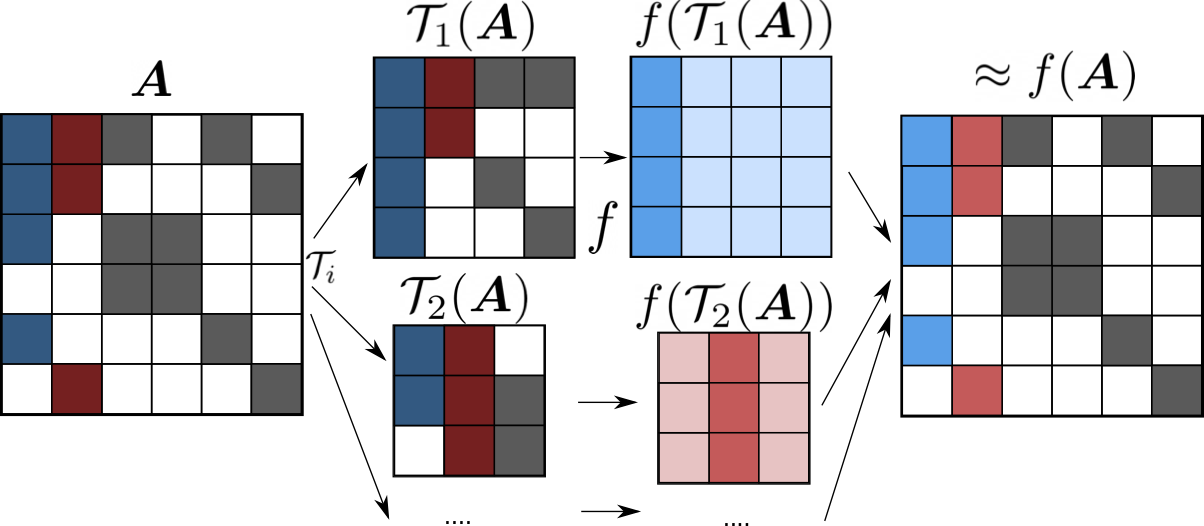
At its core, the method computes an approximated matrix function on a very large, sparse matrix, which is a key operation in quantum mechanical linear-scaling electronic structure computations. To this end, the method divides the eventually resulting density matrix, which is a huge sparse matrix, into many much smaller, but dense, submatrices, on which the matrix function is evaluated, and assembles these intermediate solutions into a global solution. As all evaluations of the matrix functions on the submatrices are independent, the method avoids communication and is extremely scalable. Since the submatrices are small and dense (order of a few thousands rows/columns) performing linear algebra on those matrices reaches close to peak performance for GPUs. The submatrix method preserves, i.e. enforces, the sparsity patterns of the original matrix. Hence, the method introduces an approximation error, whose magnitude is acceptable for this application as shown in the publications. Additionally, the application can be made tolerant for low-precision computing by compensating the introduced errors with a Langevin-type equation for the atomic motion. This opens the way to the use of mixed-precision computing using tensor cores, which translates to one magnitude more performance than double or single precision arithmetic.
Record-size simulation on the JUWELS Booster supercomputer
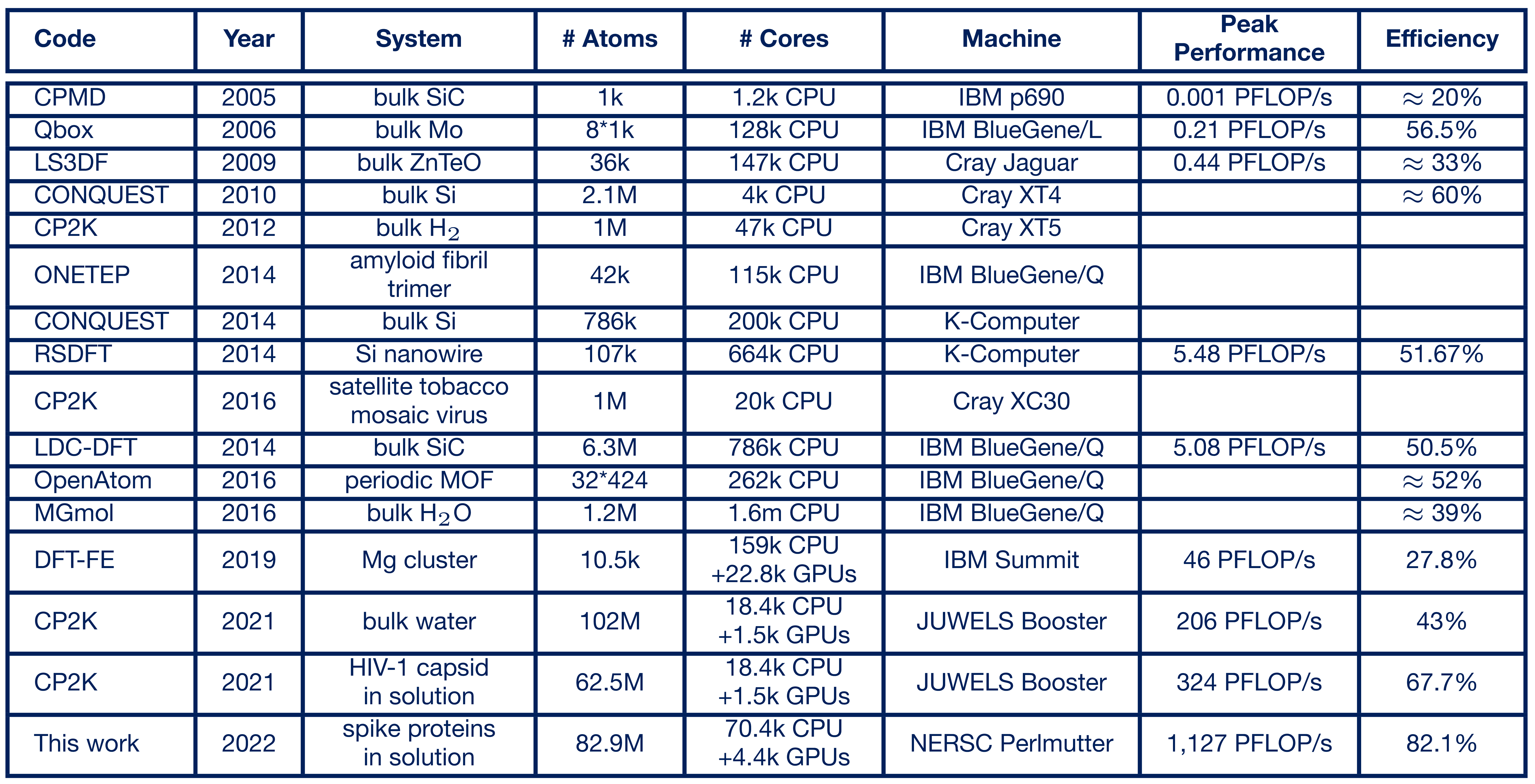
To evaluate the submatrix method, it was integrated into the popular open-source quantum chemistry program CP2K (https://cp2k.org/). There it is used within the xTB method for solving electronic structure problems, which are by far the largest computational time component for ab initio molecular dynamics simulations. In 2021, the Paderborn scientists performed simulations of the HIV virus with up to 102 million atoms on what was then Europe's fastest supercomputer (now ranked 8th worldwide), the "JUWELS Booster" at the Jülich Supercomputing Centre, setting a record for the largest electronic structure-based ab initio molecular dynamics simulation. This achieved a computational performance of 324 petaflop/s in mixed-precision floating-point arithmetic and an efficiency of 67.7% of the theoretically available computational power, which is outstanding for this application domain (https://doi.org/10.1016/j.parco.2022.102920).
Performance world record on Perlmutter supercomputer at NERSC
Since the record simulation in Jülich, the method has been further optimized to increase the efficiency of using the GPU hardware accelerators, in particular, by combining suitable submatrices for the GPU, such that the GPU can operate even closer to peak performance. To practically test the exascale capability of the method, the team was able to secure early access to the "Perlmutter" supercomputer at NERSC, currently ranked as number five on the Top 500 list, which has sufficient computing resources to break the exascale barrier for mixed-precision arithmetic.
In April 2022, the team was able to report completion! In a simulation of Covid-19 spike proteins, the exaflop barrier was broken for the first time in a real-world scientific computing application using 4,400 GPU accelerators, and 1.1 exaflop/s in mixed precision arithmetic was achieved in the computation-time critical part of the application (https://doi.org/10.48550/arXiv.2205.12182).
To classify this breakthrough, we can consider that a single simulation step for 83 million atoms takes 42 seconds, performing about 42 * 1.127 * 1018 = 47 * 1018 floating-point operations. Excluding memory requirements, such a calculation step would have taken ~47,000s or about 13 hours with the first petaflops-class system, Roadrunner from 2008, and about 1.5 years with the first teraflops-class system, ASCI Red from 1997.
Next Steps
With this success, the topic is far from exhausted for the groups involved, and the team is already working on next steps. The gold standard for atomistic simulations in chemistry and solid-state physics is the density functional theory method. The team is very confident that it can apply the submatrix method to density functional theory.





{kind=link}
{kind=link}
{kind=link}